Zusammenhang Hufrehe und Thiaminpyrophosphat
Die Tatsache, dass es bei meinem Pony bisher jedesmal an etwas gelegen hat, das den Vitamin B1-Haushalt gestört hat, dass sie Hufrehe bekommen hat, hat mich zu der Überlegung angeregt, ob es sein könnte, dass der Typ des Rehepferdes, der zum equinen metabolischen Syndrom neigt, möglicherweise zu wenig Thiaminpyrophosphat im Körper hat und aus diesem Grund schneller als seine kerngesunden Kollegen bei gefährlichem Futter zu Hufrehe neigt, also früher kollabiert. Und was ich jetzt gefunden habe, scheint diese Vermutung zu bestätigen, jedenfalls in etwa. Es scheint ein bisschen anders zu sein. Vermutlich genetisch bedingt neigen laut Versuchen alle Lebewesen, die zum metabolischen Syndrom neigen dazu, Glucose und Glykogen zu sparen und statt dessen vermehrt Fettsäuren für die Energiegewinnung zu nutzen, weshalb sie auch eine verminderte Aktivität der Lipoproteinlipase aufweisen .. auch das sage ich ja schon lange, dass ich das vermute, stimmt also auch.
Es ist laut dieser Versuche vermutlich nicht sinnvoll, künstlich durch etwas, was diesen Kreislauf verändert, was zu ändern .. es ist sinnvoller, sich desse bewusst zu sein und so zu leben, wie es am besten für ein Lebewesen ist, das zum metabolischen Syndrom neigt.
Ich würde fast sagen, man sollte nicht sagen, das ist eine Krankheit, sondern ein individueller Zustand, dem man Rechnung tragen sollte und beim Rehepferd heißt das keine Calcium unc Vitamin B1-Räuber und nicht zu viel Zucker, wohingegen Fett für diesen Typ eher nicht schädlich ist, denn das kann er ja gut verdauen und hortet es nicht, sondern er hortet zu viel Zucker und macht daraus dann eben auch Fett.
Dazu möchte ich einmal auflisten, wie das bei Chiwa ausschaut:
1. zu viel Weißbrot, schon als Jungpferd ... bei Chiwa Kolik, bei den anderen beiden nicht
2. zu viel Sumpfschachtelhalm ... bei Chiwa ganz schlimm Hufrehe, bei Reno ein leichtes Hufgeschwür, bei Nixe gar nichts
3. zu viele Eicheln, bei Chiwa Hufrehe, bei Reno gar nichts
4. zu viel Weißklee, bei Chiwa fast Hufrehe, das mehrmals (Speckhals, verspannte Muskeln), bei Reno und Prima nichts zu merken
5. Sohle zu dünn ausgeschnitten ... bei Reno mal ein kleines Hufgeschwür, bei Chiwa ganz schlimme Hufgeschwüre, etliche, so dass sie fast daran eingegangen wäre ... erinnert mich an die im folgenden Artikel beschriebenen total empfindlichen diabetischen Füße
6. Schimmel in Heulage ... 2007 bei Chiwa schlimmer Speckhalt, bei Prima leicht zu merken, sind dann geflüchtet und haben den Stall gewechselt - 2013 dann weil wir nicht geflüchtet sind, bei Chiwa Hufrehe, bei einem anderen Pferd Kolik, das aber das Cushing-Syndrom hatte ...die anderen Pferde haben es verkraftet, auch wenn es sicher für keines gesund gewesen ist.
Alle diese Dinge außer Punkt 5 sind von Futter ausgelöst worden, das auf irgendeine Weise das Vitamin B1 hemmt.
Vitamin B1 wird im Körper zu Thaminpyrophosphat umgewandelt und sorgt dann dafür, dass das Pyrovat umgewandelt wird und über drei weitere Schritte als Acetyl-CoA in den Citratzyklus eingeschleust werden kann. Wenn das nicht zuverlässig klappt, neigt ein Lebewesen, also auch ein Pferd, dazu, ständig im Körper wieder zu viel Glucose aufzubauen, es überzuckert also, egal was es frisst, ohnehin schon von innen sehr leicht, was man mit viel Bewegung und sehr guter Überlegung beim Füttern aber meistens noch im Griff behalten kann.
Aber jedes Gift wie in das Sumpfschachtelhalm, zu viel Gerbsäure in Klee und Eicheln, Schimmel in Heulage, Heu oder auch anderen Dingen sowie zu viele Kohlenhydrate, die das Pferd aufgrund seiner schiefen Stoffwechsellage auch nicht verträgt, führt wesentlich schneller zu Hufrehe als bei einem gesunden Pferd.
Es ist anzumerken, dass es die Menge macht. Alles, was bei einem Rehepferd zu Hufrehe führt, tut es auch bei anderen Pferden. Diese Pferde vertragen von diesen Sachen lediglich größere Mengen als Pferde, die das equine metabolische Syndrom haben.
Nun mal einige Quellen, die aussagen, dass ich tatsächlich recht habe.
Daraus diese Textstelle:
Stark vereinfacht dargestellt, ist die Transketolase ein „Entgiftungsenzym“.
Allerdings benötigt dieses für seine Tätigkeit
den Kofaktor Thiaminpyrophosphat (TPP, aus Vitamin
B1). Mangelt es daran, wie es bei Diabetikern zumeist der
Fall ist, ist die Transketolase-Aktivität erheblich beeinträchtigt.
Bei Typ-1-Diabetikern sind im Vergleich zu Stoffwechselgesunden
die Thiamin-Plasmaspiegel um durchschnittlich
76 Prozent erniedrigt und bei Typ-2-Diabetikern um
75 Prozent.5 Hauptgrund hierfür ist eine erhöhte Thiaminausscheidung
aufgrund von diabetischen Nierenschäden.
Aus dieser Erkenntnis heraus und den zahlreichen Hürden
einer normnahen Einstellung des Blutzuckers ergibt sich der
logische Schluss, den Thiaminmangel eines Diabetikers zu
beheben, um glukotoxischen Wirkungen und damit assoziierten
diabetischen Folgeerkrankungen entgegenzuwirken.
Dafür sprechen außerdem die zahlreichen Probleme, in der
Praxis eine normnahe Blutzuckereinstellung zu erreichen.
Diese Seite empfiehlt dann ein Medikament, aus dem nach deren Angaben leichter als einfach durch eine gesunde Ernährung das Thiaminpyrophosphat gebildet werden können soll.
Der nächste Link ist aus einer Uni-Studie:
Darum geht es in dieser Studie; ich zitiere mal kurz:
Da im Rahmen dieser Arbeit sowohl Tiere mit einem ungestörten als auch
einem gestörten Glukosestoffwechsel verwendet wurden, wird zum besseren
Verständnis deren Intermediärstoffwechsels (gehungert, Glukosebelastung) zunächst
auf die Physiologie und Pathophysiologie eingegangen. Im Anschluss wird die
Bedeutung des Pyruvatdehydrogenase (PDH)-Komplexes als zentrales
Schlüsselenzym der Glukoseoxidation und als mögliches Ziel für pharmakologische
Eingriffe aufgezeigt
Das nächste Stück Text übernehme ich auch mal als Zitat:
Rolle des Pyruvatdehydrogenase-Komplexes
Der mitochondriale Pyruvatdehydrogenase-Komplex katalysiert die irreversible
dehydrierende Decarboxylierung von Pyruvat unter Bildung von Kohlendioxid (CO2),
Acetyl-CoA und NADH.
Unter gefütterten Bedingungen ist der Komplex enzymatisch aktiv und erzeugt
Acetyl-CoA, das entweder im Krebs-Zyklus vollständig zu CO2 und H2O abgebaut
wird oder als Substrat für die Fettsäuresynthese genutzt wird (Randle, 1986). Im
gehungerten Zustand ist der Komplex inaktiv, um Pyruvat und Laktat für die
Glukoneogenese aufzusparen (Randle, 1986). Der PDH-Komplex verbindet somit als
Knotenpunkt die Glykolyse und den Citratzyklus (Harris et al., 2002) und fungiert als
wichtiges Schlüsselenzym der Blutglukosehomöostase.
Der PDH-Multienzymkomplex (Abb. I-1) besteht aus 3 katalytischen
Komponenten (E1, E2, E3) (Patel u. Korotchkina, 2001). Die Pyruvatdecarboxylaseeinheit
(E1) ist ein Tetramer der Zusammensetzung α2β2 (Reed, 1974), das die
geschwindigkeitsbestimmende Teilreaktion des Komplexes katalysiert, wobei
Thiaminpyrophosphat (TPP) als Coenzym zur Decarboxylierung von Pyruvat und zur
reduktiven Acetylierung der Lipoatgruppen (Lip-S2) dient, die kovalent an die
Lipotransacetylase (E2) gebunden sind. Der Acetylrest wird mit dem Coenzym A
unter Bildung von Acetyl-CoA abgespalten.
Das könnte auch wichtig sein:
Bislang unerforscht blieben Untersuchungen zur Änderung der PDH-Aktivität
während eines oGTT. Aussagen über den Aktivitätszustand der PDH werden meist
nur indirekt über die Western- oder Northern-Blotting-Analyse der PDK oder PDP
gemacht, ohne die Aktivität des Enzym-Komplexes zu bestimmen. Um die
Regulation des PDH-Enzymkomplexes während einer oralen Glukosebelastung zu
Einleitung
22
studieren, wurde die PDH-Aktivität aus dem Leber- und Muskelgewebe zu definierten
Zeitpunkten gemessen.
Die Ergebnisse zeigen, dass es ein Unterschied ist bei diesem PDH-Kompex, wozu auch Thiaminpyrophosphat (TPP) gehört, ob die Ratten gesund und schlank waren oder zum metabolischen Syndrom neigten.
III.2.4.3 Biochemische Parameter
Nach den in vivo Studien wurde die Regulation des PDH-Komplexes ex vivo
anhand der PDH-Aktivität und der PDK4 Expression im Leber- und Muskelgewebe
untersucht. Die PDH-Aktivität und die PDK4-Proteinmenge wurden zunächst im
Grundzustand (gefüttert vs. gehungert bzw. schlank vs. obese) und anschließend
während des oGTT bestimmt.
III.2.4.3.a PDH-Aktivität
Die Enzymaktivität im Lebergewebe (Abb. III-35, A) gefütterter Tiere (schlank:
3,0 mU/mg, obese: 2,7 mU/mg) lag um den Faktor 1,8 (schlank) bzw. 2,2 (obese)
höher als bei den gehungerten Tieren (schlank: 1,7 mU/mg, obese: 1,2 mU/mg).
Die Aktivitäten im Muskelgewebe (Abb. III-35, A) bewegten sich insgesamt auf
einem höheren Niveau. Der Substratumsatz war bei gefütterten Tieren in der
Muskulatur ca. zweifach höher als in der Leber (schlank: 8,2 mU/mg, obese: 6,9
mU/mg).
Zusätzlich wurde die Gesamtaktivität (Abb. III-35, B) durch Zugabe von λ-
PPase (siehe Kapitel II.2.2.a) bestimmt. Hier zeigte sich, dass im Lebergewebe die
PDH-Aktivität bei obesen Tieren ca. 20% über der der schlanken Ratten lag,
während in der Muskulatur zwischen diesen Gruppen keine Unterschiede zu
erkennen waren.
.......
Mit Hilfe der Gesamtaktivität ließ sich der prozentuale Anteil der PDH-Aktivität
(Abb. III-36) berechnen.
Es zeigte sich, dass sich die Werte der Leber-PDH obeser Ratten aufgrund
der höheren Totalaktivität stärker von denen der schlanken Tiere unterschieden.
Für beide Gewebe ergab sich, sowohl bei schlanken als auch bei obesen
Tieren, ein Aktivitätsverhältnis von 2:1 für gefütterte vs. gefastete Stoffwechsellage.
In der Leber war unter Fütterung die PDH lediglich zu 25% (schlank) bzw.
14% (obese) aktiv, wohingegen die Anteile im Muskel bei 79% (schlank) bzw. 73%
(obese) lagen
.....
PDH-Aktivität
Die Enzymaktivität (siehe Kapitel III.2.2.a) im Lebergewebe (Abb. III-62, A) bei
obesen und schlanken Tieren war unter gefütterten Bedingungen mit 1 mU/mg
identisch. In der Muskulatur (Abb. III-62, A) lagen die PDH-Aktivitätswerte bei
gefütterten, schlanken und obesen Tieren um einen Faktor 2 höher als nach
Nahrungskarenz. Die Aktivität betrug gefüttert bei schlanken Tieren 8,4 mU/mg bzw.
6,6 mU/mg (obese) und gehungert 4,0 mU/mg bzw. 3,2 mU/mg (obese).
Zusätzlich wurde die Gesamtaktivität (Abb. III-62, B) durch Zugabe von λ-
PPase (siehe Kapitel II.2.2.a) bestimmt.
Hier zeigte sich, dass die „totale Aktivität“ der PDH obeser Tiere sowohl im
Lebergewebe als auch in der Muskulatur im Vergleich zu den schlanken Tieren
signifikant erhöht war
...
Mit Hilfe der Gesamtaktivität ließ sich der prozentuale Anteil aktiver PDH (Abb.
III-63) berechnen.
Es zeigte sich, dass der Anteil aktiver PDH bei den schlanken ZDF-Ratten
signifikant höher lag als bei den obesen Ratten, während bei Betrachtung der
spezifischen Aktivität (Abb. III-62, A) der Unterschied zwischen den beiden Gruppen
kaum zu sehen war.
In der Leber war unter Fütterung die PDH lediglich zu 16% (schlank) bzw.
10% (obese) aktiv, wohingegen die Anteile im Muskel bei 73% (schlank) bzw. 44%
(obese) lagen
...
DCA ist sicher ein Medikament .. ich werde das nachher anhand der Kürzel, die immer in solchen Arbeiten zu finden sind, raus suchen.
...
Obwohl nach einmaliger DCA-Behandlung obeser ZDF-Ratten eine
Steigerung der PDH-Aktivität festgestellt werden konnte (Abb. III-74), die zu einer
Senkung der Laktatspiegel (Abb. III-70, B) führte, wurde für den gewählten
Versuchszeitraum keine blutglukosesenkende Wirkung beobachtet (Abb. III-69, B).
Daher wurde der Versuch auf eine Dauer von acht Stunden ausgedehnt, um zu
untersuchen, ob der Blutglukoseabfall möglicherweise verzögert auftritt
....
Um den Zusammenhang zwischen Kohlenhydrat- und Fettstoffwechsel beim
Übergang vom gefütterten in den gehungerten Zustand zu verdeutlichen, wurde für
die Kontrollen (Abb. III-79, A/B) und die behandelten Gruppen (Abb. III-79, C/D) die
KHO und die FO jeweils in einer gemeinsamen Graphik dargestellt.
Der Verlauf der KHO und der FO ergab schematisch gesehen das Bild einer
„Schere“, in deren Schnittpunkt die Umstellung von Glukose- auf Lipidoxidation
deutlich wird. Nach DCA Behandlung näherten (Abb. III-79, C) sich die beiden
Kurven der KHO und der FO, bzw. liefen parallel (Abb. III-79, D).
...
......
Und auch wenn das jetzt nichts über das Thiamin oder Thiaminpyrophosphat ist, sondern
etwas, das ich schon seit Jahren immer wieder sage, auch die Lipoproteinlipase muss bei Lebewesen, die zur Insulinresistenz und dem metabolischen Syndrom neigen, weniger aktiv sein .. und das ist bei diesem Versuch bestätigt worden: Lest mal selbst:
....
Der Hypertriglyzeridämie (Abb. III-48, B) beim Typ 2 Diabetes liegt eine
Vermehrung der VLDL-Spiegel zugrunde (Grundy, 1997). Die Erhöhung dieser
Lipoproteine sehr geringer Dichte liegt zum einen in der verminderten Aktivität der
Lipoproteinlipase begründet, zum anderen findet bei Insulinresistenz eine
verminderte Suppression der VLDL Sekretion in der Leber statt (Malmström et al.,
1997).
.....
Das ist auch wichtig:
Für schlanke und obese ZDF-Ratten konnte gezeigt werden, dass die
Senkung der Blutglukose auf den Wirkmechanismus einer gesteigerten PDH-Aktivität
zurückzuführen ist. Jedoch zeigten schlanke und obese Tiere unter der Behandlung
ein unterschiedliches Muster.
Bei den insulinsensitiven Ratten sank zwei Stunden nach der ersten
Applikation (DCA 1x: Abb. III-69, A) die Blutglukose, während bei den obesen Tieren
innerhalb der ersten vier Stunden (Abb. III-69, B), trotz gesteigerter PDH-Aktivität
(Abb. III-73) keine Reaktion auf die Behandlung zu beobachten war. Anders verhielt
es sich bei Laktat (Abb. III-70); unmittelbar nach Applikation des PDK-Inhibitors fielen
die Laktatwerte sowohl in der schlanken als auch in der obesen Gruppe im Vergleich
zur Kontrolle signifikant ab. Aufgrund dieser Ergebnisse scheint Laktat für ein in vivo
Monitoring der PDH, bezüglich unmittelbarer Aktivitätsänderungen in einem kurzen
Versuchszeitraum, der sensiblere Blutparameter zu sein.
Da bei der zweimaligen Applikation (DCA 2x) die Substanz bereits am Vortag
gegeben wurde, lagen die Blutglukose- und Laktatwerte jeweils bei einem tieferen
Wert. Diese Beobachtung lässt den Schluss zu, dass die Inaktivierung der PDH, um
nach dem Nahrungsentzug Glukose zu sparen, bis zu einem gewissen Grad
verhindert werden konnte.
Des Weiteren kann aus den Daten gefolgert werden, dass die Wirkung des
Inhibitors über mindestens 18 Stunden anhielt.
Bei den jeweils zweimal behandelten Gruppen konnte die zweite DCA Gabe
keine weitere Blutglukosesenkung herbeiführen bzw. Laktat nur noch in geringem
Maße senken. Möglicherweise ist nach der ersten Behandlung der pharmakologische
Effekt auf die PDH Aktiviät nicht mehr weiter zu steigern.
Die Ergebnisse der Glykogenbestimmungen (Abb. III-71, 72) zeigen den
Zusammenhang zwischen der gesteigerten PDH-Aktivität und der Abnahme der
Glykogenvorräte. Diese Abnahme konnte in der Leber und im Muskel der obesen
ZDF-Ratten beobachtet werden und war abhängig von der Anzahl der Applikationen.
Unter DCA Behandlung konnte bereits nach einer einmaligen Dosis die
Enzymaktivität im Leber- und Muskelgewebe (Abb. III-73) in etwa verdoppelt werden.
Bezeichnet man den prozentualen Anteil nicht aktiver PDH in den Geweben der
Kontrolle als sogenanntes therapeutisches Fenster, so war nach der
pharmakologischen Intervention noch ein Spielraum von etwa 20% (Muskel) bzw.
50% (Leber) für eine weitere Aktivitätssteigerung zum theoretisch möglichen Wert
von 100% vorhanden (Abb. III-74). Die begrenzte Steigerung der PDH-Aktivität,
insbesondere in der Leber, ist möglicherweise Ausdruck der Unspezifität von DCA
als PDK-Inhibitor. Weitere Steigerungen wären durch spezifische Inhibitoren möglich
(Mayers et al., 2005), wobei eine vollständige Dephosphorylierung aufgrund der
vielfältigen Interkonvertierung des Enzymkomplexes nur als theoretisches Maximum
angesehen werden kann.
Da bei der obesen Gruppe nach einmaliger Gabe von DCA kein
Blutglukoseabfall festzustellen war, obwohl die sinkenden Laktatspiegel eine PDHAktivierung
vermuten ließen und diese auch bestätigt werden konnte, wurde der
Versuchszeitraum auf 8 Stunden ausgedehnt, um diesen Sachverhalt zu eruieren.
.....Ich bin nur ein Laie mit gewissen Kenntnissen in der Ernährungslehre, würde aber daraus schließen, dass die Neigung zu Hufrehe sowohl bei Pferden mit EMS als auch EPSSM zusammenhängt, denn die Rehepferde, die zu EPSSM neigen, haben nachgewiesen zu große Glykogenspeicher.
Das sind hier zwar Ratten und weder Menschen noch Pferde, in dem Fall allerdings scheint dieser Komplex dafür zu sorgen, dass Glucose und Glykogen sozusagen für schlechte Zeiten gespart werden.
Dieser Tierversuch hat wohl den Grund, dass sie an den Ratten testen wollten, ob es Sinn macht, bei Menschen zu versuchen, mehr Glucose über den Citratzyklus zu verstoffwechseln, um so die Insulinresistenz zu beseitigen, also ob das Sinn mache.
Ich suche ja was anderes, nämlich eigentlich nur, ob es sein kann, dass Pferde, die zu Hufrehe neigen, möglicherweise eben auch dazu neigen, Glucose und Glykogen aufzuspeichern statt es zu verstoffwechseln und deshalb besonders empfinglich auf Vitamin B1-Räuber reagieren und ich würde sagen, diese Versuche lassen vermuten, dass das so sein wird.
Was der Autor dieser PDF sagt, finde ich sehr wichtig und möchte es auch noch übernehmen:
Jedoch darf nicht außer Acht gelassen werden, dass die Hyperglykämie nur
eine Dekompensation im komplexen Stoffwechsel des Typ 2 Diabetes darstellt und
dass pharmakologische Eingriffe im Glukosestoffwechsel wiederum andere
biochemische Pfade beeinflussen können, wie zum Beispiel den Lipidmetabolismus.
Ich sage ja auch immer, alles was man an chemischen Eingriffen umgehen kann, sollte man umgehen, wenn es sich durch eine Lebensweise, die auf die spezifischen Stoffwechselbedingungen eines Lebewesens so eingehen, dass ihm nichts passiert, umgehen lässt.
http://de.wikipedia.org/wiki/Dichloressigs%C3%A4ure
Dieses Mittel DCA hat sich nicht unbedingt als gesund erwiesen, wie man Wikipedia entnehmen kann.
FO ist die Fettoxidation .. die ist in den Teilen, die ich zitiert habe, auch erwähnt.
KHO ist die Kohlenhydratoxidation.
Lebewesen mit Insulinresistenz sparen also Kohlenhydrate aufgrund ihrer genetischen Veranlagung (die Ratten waren genetisch verändert worden) und holen sich die benötigte Energie eher aus der Fettoxidation, wenn sie können.
Vermutlich würden sie übrigens noch dicker werden, wenn man sie mit Medikamenten behandeln würde, die dazu führen, dass dieser Stoffwechselweg mehr über den Citratcyklus liefe.
http://de.wikipedia.org/wiki/Pyruvatdehydrogenase-Komplex
Daraus nur mal die wichtigsten Sachen, damit verständlich ist, worum es da geht.
Die Reaktion
Bei der oxidativen Decarboxylierung wird vom Pyruvat (C3) Kohlenstoffdioxid (CO2)
abgespalten und ein NADH gewonnen. Dabei wird eine energiereiche
Thioesterbindung zwischen Coenzym A und dem Acetatrest gebildet, so dass
Acetyl-CoA
entsteht. Die Energie hierfür stammt aus der Decarboxylierung. Die
Umwandlung von Pyruvat zu Acetyl-CoA ist unter physiologischen
Bedingungen irreversibel.
Teilschritte
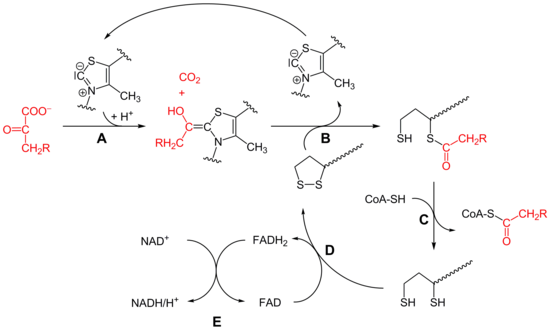
- Die Decarboxylierung von Pyruvat erfolgt mit Hilfe der Pyruvatdehydrogenase (E1) des Pyruvatdehydrogenase-Komplex (A). Bei dieser katalysierten Reaktion ist Thiaminpyrophosphat (TPP) die prosthetische Gruppe und bildet eine Atombindung mit Pyruvat. Das Reaktionsprodukt ist Hydroxyethyl-TPP und CO2. Diese Hydroxyethylgruppe wird zu einer Acetylgruppe oxidiert und von Liponamid übernommen, so dass eine energiereiche Thioesterbindung, S-Acetylliponamid (B),
entsteht. Liponamid ist an der Transacetylase-Untereinheit kovalent
gebunden. Die Disulfidgruppe des Liponamids wird bei dieser Reaktion zur
Disulfhydrylform reduziert.
- Der Acetylrest von Acetylliponamid wird auf Coenzym A übertragen, somit entstehen Acetyl-CoA und Dihydroliponamid (C). Dies wird von der Dihydrolipoyl-Transacetylase (E2) katalysiert. Formal erfolgt bei dieser Reaktion eine Umesterung, wodurch die energiereiche Thioesterbindung erhalten bleibt.[9]
- Dihydroliponamid wird durch die Dihydrolipoyl-Dehydrogenase (E3)-Untereinheit zu Liponamid regeneriert. Dabei wird ein kovalent gebundenes FAD zu FADH2 reduziert (D), welches durch die Reduktion von NAD+ wieder regeneriert wird (E).
Die Übertragung von Elektronen findet normalerweise in umgekehrter
Richtung von NADH zu FAD statt. Das Elektronenübertragungspotential FADs
ist durch seine kovalente Bindung mit dem Protein aber ausreichend
erhöht, so dass die Reaktion ablaufen kann.[9]
Somit ergibt sich folgende Gesamtreaktion:

Durch die Generierung von Acetyl-CoA aus Pyruvat wird eine Verbindung
zwischen der Glykolyse und Citratzyklus hergestellt. Das entstandene
Acetyl-CoA kann dann mit
Oxalacetat durch die
Citratsynthase weiter zu
Citrat umgesetzt werden. Das NADH/H
+ kann durch die
Atmungskette wieder reoxidiert werden.
Essentialität von Vitamin B1 und Mangel
Der Pyruvatdehydrogenase-Komplex ist, gemäß der beschriebenen
Reaktion, für alle (netto-) Energiegewinnung aus Kohlenhydraten (im
Gegensatz zu
Fetten) notwendig. Mit dem Anteil von
Vitamin B1
(Thiamin) ist hierzu auch ein Vitamin nötig, also ein Stoff der von
außen zugeführt werden muss. Es gibt einen erhöhten Bedarf für Thiamin
bei stark erhöhter Kohlehydratzufuhr. Bei normaler gesunder Ernährung
ohne Alkoholkonsum ist eine zusätzliche Thiaminzufuhr nicht notwendig.
[10][11][12][13][14]
Regulation
Die Endprodukte Acetyl-CoA und auch NADH können zu einer Hemmung des
Pyruvatdehydrogenase-Komplexes führen (Produkthemmung). Darüber hinaus
wird der Komplex auch durch zwei Modifikationen reguliert. Hierbei
katalysieren eine Pyruvatdehydrogenase-Kinase (PDK) und eine
Phosphopyruvatdehydrogenase-Phosphatase (PDP) die reversible
Phosphorylierung des cytosolischen PDC.
[15] In Säugern werden drei, in Pflanzen zwei hochkonservierte
Serinreste der E
1-Untereinheit durch die PDK unter
ATP-Verbrauch
phosphoryliert. Dies bewirkt eine komplette Inaktivierung der PDC. Die
Phosphatase macht die Phosphorylierung wieder rückgängig und aktiviert
damit den Gesamtkomplex.
Beim Menschen wird die PDP durch
Calcium- sowie
Magnesiumionen stimuliert.
[16] Eine Steigerung des Calciumspiegels kann auch von
α-Sympathomimetika und
Vasopressin hervorgerufen werden. Die PDK wird dagegen von Acetyl-CoA und NADH stimuliert, während Pyruvat,
ADP
und Calciumionen einen hemmenden Effekt haben. In Pflanzen ist die
Aktivität der Kinase höher als die der Phosphatase, so dass sie dort
noch zusätzlich reguliert werden muss. Hierbei aktiviert
Ammonium (NH
4+) die PDK, während Pyruvat und ADP diese hemmen.
Das macht dann auch verständlich, warum es so schädlich ist, wenn Pferdefutter Stoffe wie die Oxalsäure enthält, die zu viel Calcium binden.
LG
Renate